Protein folding
- Analisi di dinamica di molecole organiche
- Studio di interazione tra diverse molecole organiche
- Studio di molecole di interesse farmaceutico (determinazione delle affinità tramite analisi software)
- Ingegnerizzazione di molecole con particolari funzioni biologiche
- Determinazione di dinamica di molecole patologiche

Qui è possibile vedere in azione un semplice simulatore sotto Java che è stato sviluppato da Proteobiochip.
Java è un linguaggio interpretato e non è veloce come il linguaggio C ma permette di vedere le possibilità del simulatore di dinamica
di molecole organiche.
Nel simulatore sotto Java è possibile simulare solo delle semplici molecole.
Per cominciare viene esposto un piccolo Help per iniziare.
Successivamente un manuale più dettagliato ed infine il link per aprire il simulatore sotto Java.
Simulatore di forma 3D dinamica delle proteine
Interfacce grafiche:

Help per iniziare
Per poter utilizzare questo simulatore, per ora e' necessario disabilitare il sistema Java security,
non mi rendo responsabile di un'uso improprio, anche se da parte mia posso dire che il software non e' soggetto a virus e non puo' recar danni al vostro sistema.
Caricare dalla finestra "Genoma" un amminoacido (es.: Fenilalanina)
(P.S. Scorrendo la finestra in cui compare azoto acqua metano etilene ...)
Dalla finestra "FinestraGrafica" premere PLAY
Selezionare a scelta i parametri di visualizzazione (Nomi,Versori,Orientamenti,DirezioneLegame,DirezioneOrientamento)
I parametri: (Distanza,Carica,Debole,Angoli)
sono solo per utenti esperti e servono per la diagnosi di funzionamento del software
Per fermare il processo premere STOP
Manuale d'uso
Manuale per il programma di simulazione dinamica tridimensionale di proteine.
Creato da Barbieri Francesco, laureato in Ingegneria Elettronica dell'università di Padova.
Il simulatore è composto di due finestre separate per garantire una più ampia area di lavoro per la
visualizzazione grafica.
Queste due finestre sono: Genoma e FinestraGrafica.
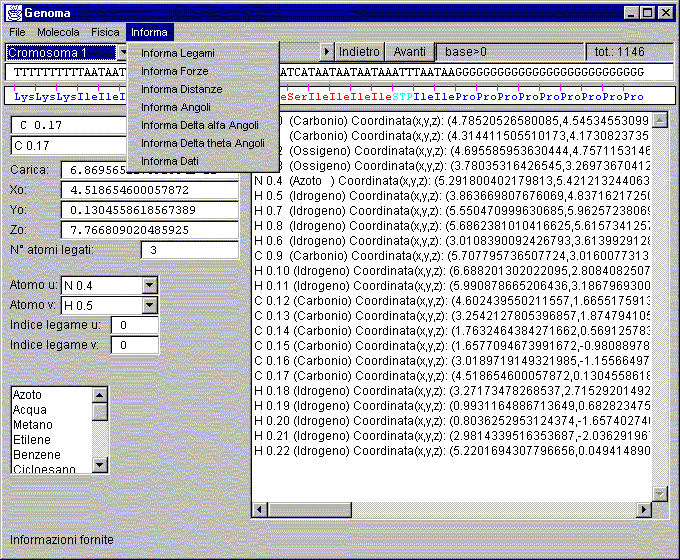
Finestra: Genoma
Finestra: FinestraGrafica
Finestra Genoma
In ordine di apparizione sullo schermo:
Descrizioni:
Menù:
File:
Molecola:
Fisica:
Informa:
Dim Font:
Load:
Carica i cromosomi dal disco.
Salva:
Salva la modifica fatta al cromosoma sul disco.
Per fare le modifiche, è sufficente scrivere sopra alla sequenza esistente.
Carica sequenza:
Carica la sequenza dal file di tipo SEQ e la converte il formato compresso DNA.
Questo formato è supportato solamente da questo software.
Nuova Molecola:
Azzera tutte le impostazioni attuali delle molecole.
Aggiungi Atomo:
Aggiunge l'atomo con le caratteristiche selezionate nell'area inferiore sinistra.
Salva Modifica:
Salva la modifica fatta all'atomo.
Quest'ultima si può fare agendo direttamente sui parametri visualizzati nell'area inferiore sinistra.
Cancella Atomo:
Cancella l'atomo selezionato nell'area inferiore sinistra.
Aggiungi Legame u-v:
Aggiunge il legame con le caratteristiche assegnate nell'area inferiore sinistra.
Elimina Legame:
Elimina il legame selezionato nell'area inferiore sinistra.
Salva su disco:
Salva su disco la molecola (o le molecole) con le caratteristiche calcolate.
Il formato è di tipo MOL ed è supportato solamente da questo programma.
Carica da disco:
Carica il file di tipo MOL dal disco il quale contiene le caratteristiche precalcolate di una o più molecole.
Il formato è di tipo MOL ed è supportato solamente da questo programma.
Distribuisci Carica:
Azione di distribuzione della carica che si svolge comunque in modo automatico alla fase di lancio del processo di calcolo.
Assesta distanze:
Esegue un passo di assestamento delle distanze relative tra atomi.
Informa Legami:
Restituisce al pannello di visualizzazione dei dati le informazioni relative al tipo di legame.
Informa Forze:
Restituisce al pannello di visualizzazione dei dati le informazioni relative alle forze sugli atomi,
scrivendone sia modulo che vettore.
Informa Distanze:
Restituisce al pannello di visualizzazione dei dati le informazioni relative alle distanze relative tra atomi,
partendo dall'atomo selezionato nell'area inferiore sinistra.
Informa Angoli:
Restituisce al pannello di visualizzazione dei dati le informazioni relative agli angoli tra gli atomi legati all'atomo
selezionato nell'area inferiore sinistra.
Informa Delta alfa Angoli:
Restituisce al pannello di visualizzazione dei dati le informazioni relative alla variazione dell'angolo di flessione
rispetto alla posizione di equilibrio determinata dall'orientamento dell'atomo (visualizzabile selezionando gli orientamenti nella finestra grafica).
Informa Delta theta Angoli:
Restituisce al pannello di visualizzazione dei dati le informazioni relative alla variazione dell'angolo di torsione
rispetto alla posizione di equilibrio determinata dall'orientamento dell'atomo (visualizzabile selezionando gli orientamenti nella finestra grafica).
Informa Dati:
Restituisce al pannello di visualizzazione dei dati le informazioni relative a dati generici del database interno.
6:
Seleziona la dimensione 6 dei font per permetterne la visualizzazione ottimale nel vostro sistema operativo.
8:
Seleziona la dimensione 8 dei font per permetterne la visualizzazione ottimale nel vostro sistema operativo.
10:
Seleziona la dimensione 10 dei font per permetterne la visualizzazione ottimale nel vostro sistema operativo.
12:
Seleziona la dimensione 12 dei font per permetterne la visualizzazione ottimale nel vostro sistema operativo.
Scelta cromosoma:
Questa lista permette di scegliere quale cromosoma analizzare, quindi lo carica dal file specifico.
Barra scorimento:
Barra per posizionarsi su un settore specifico del cromosoma scorrendovi in modo rapido.
Indietro:
Spostamento retrocedente con un passo di una triplette.
Avanti:
Spostamento in avanti con un passo di una triplette.
Label base:
Indice della posizione sul cromosoma.
Label tot.:
Valore della dimensione complessiva del cromosoma.
Area testo per lista sequenze:
Lista della parte della sequenza caricata resa visibile.
Area testo per lista decodifica delle sequenze:
Area di decodifica delle sequenze, nella quale si visualizza in blu scuro la parte non codificante, in azzurro la tripletta di inizio e in rosso la parte codificante
(se l'inizio è visibile nell'area di visualizzazione).
Casella testo nome atomo:
Area per inserire il nome dell'atomo da inserire o inserito e selezionato.
Casella testo tipo atomo:
Area per scegliere il tipo dell'atomo da inserire o inserito e selezionato.
Casella testo lista atomi della molecola:
Area per scegliere l'atomo desiderato dalla lista di atomi memorizzati.
Casella testo carica apparente atomo:
Area per visualizzare la carica apparente dell'atomo selezionato.
Casella testo coordinata X0:
Area per visualizzare e modificare la posizione dell'atomo selezionato.
Il salvataggio della modifica si esegue dal menù.
Casella testo coordinata Y0:
Area per visualizzare e modificare la posizione dell'atomo selezionato.
Il salvataggio della modifica si esegue dal menù.
Casella testo coordinata Z0:
Area per visualizzare e modificare la posizione dell'atomo selezionato.
Il salvataggio della modifica si esegue dal menù.
Casella testo N° atomi legati:
Area per visualizzare e modificare il numero di atomi legati all'atomo selezionato.
Il salvataggio della modifica si esegue dal menù.
Casella testo Atomo u:
Area per la scelta del primo atomo da cui parte il legame.
Il salvataggio dell'aggiunta si esegue dal menù.
Casella testo Atomo v:
Area per la scelta del secondo atomo a cui arriva il legame.
Il salvataggio dell'aggiunta si esegue dal menù.
Casella testo Indice legame u:
Area per la scelta del primo indice di legame dell'atomo da cui parte il legame.
Il salvataggio dell'aggiunta si esegue dal menù.
Casella testo Indice legame v:
Area per la scelta del secondo indice di legame dell'atomo a cui arriva il legame.
Il salvataggio dell'aggiunta si esegue dal menù.
Scelta molecole non precalcolate dal database interno:
Area per la scelta delle molecole disponibili per avviarne il processo di calcolo.
E'sufficente cliccare UNA sola volta.
Area testo per comunicazioni di dati:
Area in cui si visualizzano i parametri richiesti dal menù.
Finestra FinestraGrafica
In ordine di apparizione sullo schermo:
Descrizioni:
Area grafica:
Area per la visualizzazione delle molecole.Con operazioni di drug si cambia l'angolo di vista
Play:
Avvia l'esecuzione del thread dei calcoli.
(N.B.Questo processo userà tutta la risorsa di calcolo del vostro processore
quindi è sconsigliato l'utilizzo su processori con frequenza di funzionamento overclockata, perchè
potrebbe portare il microprocessore in condizioni dannose di sovratemperatura)
Stop:
Arresta l'esecuzione del thread dei calcoli.In alcuni sistemi questa opzione non garantisce l'arresto
indeterminato. Allo stato di stop è possibile mutare il sistema dei dati per modificare le molecole.
E' sconsigliato modificare le molecole in esecuzione perchè potrebbe portare in blocco il programma.
Sblocca:
Genera uno spostamento di modulo costante e piccolo rispetto le dimensioni di lunghezza di legame medie e
di orientamento random.Questo consente di portare il sistema dei dati fuori da condizioni critiche
di errore.
Priorità processo:
Questa azione serve per assestare la priorità del processo di calcolo rispetto al processo
di interfaccia con l'utenza, permettendo di settare le condizioni ottimali per ogni tipo di piattaforma.
Questo però se usato senza cautela può bloccare la normale esecuzione del software.
Orienta:
Questo comando porta la visualizzazione grafica alle condozioni di orientamento iniziali.
Rotazioni x destrogiro e sinistrogiro:
Consente la rotazione attorno all'asse x di un angolo costante secondo il verso prescelto.
Rotazioni y destrogiro e sinistrogiro:
Consente la rotazione attorno all'asse y di un angolo costante secondo il verso prescelto.
Rotazioni z destrogiro e sinistrogiro:
Consente la rotazione attorno all'asse z di un angolo costante secondo il verso prescelto.
Distanza:
Operazione di test software:Elimina dal calcolo il parametro distanza.
Carica:
Operazione di test software:Elimina dal calcolo il parametro carica.
Debole:
Operazione di test software:Elimina dal calcolo il parametro debole.
Angoli:
Operazione di test software:Elimina dal calcolo il parametro angoli di flessione e torsione.
Nomi:
Operazione che permette di visualizzare sull'area grafica i nomi associati ai relativi atomi
Versori:
Operazione che permette di visualizzare sull'area grafica i Versori delle forze associati ai relativi atomi.
Orientamenti:
Operazione che permette di visualizzare sull'area grafica gli orientamenti associati ai relativi atomi.
Direzione legame:
Operazione che permette di visualizzare sull'area grafica i legami tra i relativi atomi, tramite dei segmenti.
Direzione torsione:
Operazione che permette di visualizzare sull'area grafica gli orientamenti di torsione tra i relativi atomi.
Link per avviare il simulatore di proteine (Protein Folding) in versione ridotta (JAVA)
Esempio Dinamica Proteine
|


