Protein folding
- Analysis of dynamics of organic molecules
- Study of interaction between various organic molecules
- Study of molecules of pharmaceutical interest (determined by software analysis of similarities)
- Engineering of molecules with specific biological functions
- Determination of pathological dynamics of molecules

Here you can see it in action just a simulator in Java that was developed by Proteobiochip.
Java is an interpreted language and not as fast as the C language, but allows you to see the possibilities of the simulator dynamics of organic molecules.
In the simulator in Java is possible to simulate only simple molecules.
For starters it is exposed to start a small Support.
Subsequently a more detailed manual and finally the link to open the simulator in Java.
3D shape simulator dynamics of proteins
Graphical user interfaces:

Help to start
In order to use this simulator, for now you need to disable the Java system security,
I am not responsible for improper use at, even if my part I can say that the software is not 'subject to viruses and can not' cause harm to your system.
Load the window "Genome" an amino acid (eg phenylalanine)
(PS you scroll the window appears in which ethylene oxide gas water ...)
From the "FinestraGrafica" press PLAY
Select your choice of display parameters (names, unit vectors, Guidelines, Link Direction, Direction Orientation)
The parameters: (Distance, Charge, Weak, Angles)
are only for advanced users and are used to diagnose operating software
To stop the process, press STOP
User Guide
Manual for the program of three-dimensional dynamic simulation of proteins.
Created by Francesco Barbieri, graduated in Electronic Engineering from the University of Padua.
The simulator consists of two separate windows to ensure a wider work area for
graphical display.
These two windows are: Genome and FinestraGrafica.
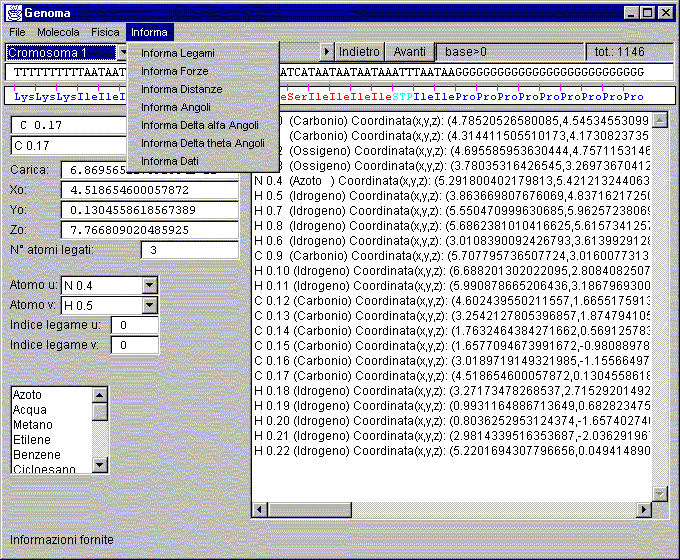
Window: Genoma
Window: FinestraGrafica
Window Genoma
In order of appearance on the screen:
Description:
Menù:
File:
Molecula:
Physics:
Information:
Dim Font:
Load:
Load chromosomes from the disc.
Save:
Save the changes made to the chromosome on the disk.
To make changes, it is sufficient to overwrite the existing sequence.
Upload sequences:
Load the sequence from the SEQ file type and converts the compressed form DNA.
This format is only supported by this software.
New Molecula:
Clear all the current settings of the molecules.
Add Atomo:
Adds the atom with the features selected in the lower left.
Save change:
Save the changes made to the atom.
This can be done by acting directly on the parameters displayed in the lower left.
Delete Atom:
Delete the selected atom in the lower left.
Add bond u-v:
Adds the bond with the characteristics assigned in the lower left.
Delete Bond:
Deletes the selected bond in the lower left.
Save to disc:
Save to disk the molecule (or molecules) with the characteristics calculated.
The format is kind of MOL and is only supported by this program.
Load from disc:
Upload files from the disc MOL type which contains the characteristics of one or more precalculated molecules.
The format is kind of MOL and is only supported by this program.
Distribute Charge:
Action of charge distribution, which takes place automatically, however, the launch phase of the process of calculating.
Set distances:
Performs one step of adjustment of the relative distances between atoms.
Information Bond:
Returns the data panel display information about the type of bond.
Information Force:
Returns to the display panel of the data and information relating to the forces on the atoms, writing both module and vector.
Information Distances:
Returns the data panel display information about the relative distances between atoms, starting from the atom selected in the lower left.
Information Angles:
Returns to the display panel of the data and information relating to the angles between the atoms bound to the atom selected in the lower left.
Information Delta alfa Angles:
Returns to the display panel of the data and information relating to the change of the angle of deflection from the position of equilibrium determined by the orientation of the atom (which is displayed by selecting the orientations in the graphics window).
Information Delta theta Angles:
Returns to the display panel of the data and information relating to the change in torsion angle relative to the position of equilibrium determined by the orientation of the atom (which is displayed by selecting the orientations in the graphics window).
Information Data:
Returns to the display panel of the data and information relating to general data in database.
6:
Select the font size 6 to allow optimum viewing in your operating system.
8:
Select the font size 8 to allow optimum viewing in your operating system.
10:
Select the font size 10 to allow optimum viewing in your operating system.
12:
Select the font size 12 to allow optimum viewing in your operating system.
Choosing chromosome:
This list allows you to choose which chromosome analysis, then load it from the file specified.
Scrolling bar:
Bar to move to a specific area of chromosome flowing quickly.
Back:
Moving with a retreating step of a triplet.
Forward:
Moving a step forward with a triplet.
Label base:
Index of position on the chromosome.
Label tot.:
Value of the overall size of the chromosome.
Text Area to sequences list:
List the loaded part of the sequence made ??visible.
Text Area for list decoding of the sequences:
Area decoding sequences, which appears in the non-coding dark blue, light blue hat-trick at the beginning and the red part of the coding
(if the start is visible in the display).
Name text box, atom:
Area to enter the name of the atom to be inserted or plugged in and selected.
Atom type text box:
Area to choose the type of the atom to be inserted or plugged in and selected.
Atoms of the molecule list box text:
Area to select the desired atom from the list of atoms stored.
Text box charge apparently atom:
Area to display the apparent charge of the atom selected.
Text Box coordinate X0:
Area to view and change the position of the atom selected.
Saving the change you from the menu.
Text Box coordinate Y0:
Area to view and change the position of the atom selected.
Saving the change you from the menu.
Text Box coordinate Z0:
Area to view and change the position of the atom selected.
Saving the change you from the menu.
N° atoms bound textbox:
Area to view and change the number of atoms bound to the atom selected.
Saving the change you from the menu.
Text Box Atom u:
Area for the choice of the first atom from which the link.
You can do the saving of addition from the menu.
Text Box Atom v:
Area for the choice of the first atom from which the link.
You can do the saving of addition from the menu.
Index text box link u:
Area for the choice of the first bond index of the atom from which the bond.
You can do the saving of addition from the menu.
Index text box link v:
Area for the choice of the first bond index of the atom from which the bond.
You can do the saving of addition from the menu.
Choosing not precalculated molecules from internal database:
Area for the selection of molecules available to start the calculation process.
Is sufficient to click only once.
Text Area for data communications:
An area in which you view the parameters required by the menu.
Window FinestraGrafica
In order of appearance on the screen:
Description:
Graphic Area:
Area for the display of molecules. With drug operations changing the angle of view
Play:
Starts the execution of the thread of the calculations.
(NB This process will use all the computing resource of your processor
so it is not recommended for use with an operating frequency of processor overclocked, because it may result in conditions harmful to the microprocessor over temperature)
Stop:
Stop the execution of the thread of the calculations. In some systems, this option does not guarantee the permanent shutdown. At the stop is possible to change the system of data to change the molecules.
It 'not recommended to change the molecules in the running because it may result in blocking the program.
Unlock:
Module generates a constant shift of the size of smaller than average bond length and orientation random. This allows the system to bring data out of critical condition
error.
Process priority:
This action serves to arrange the priority of the calculation process to the process with the user interface, allowing you to set the optimal conditions for each type of platform.
This, however, if used without caution can block the normal execution of the software.
orientation:
This command turns the display graphics to the initial orientation condozioni.
Rotations around x right and left turn:
Allows rotation around the axis x of a constant angle in the direction chosen.
Rotations around y right and left turn:
Allows rotation around the axis y of a constant angle in the direction chosen.
Rotations around z right and left turn:
Allows rotation around the axis z of a constant angle in the direction chosen.
Distances:
Operation test software: Delete the parameter distance from the calculation.
Charge:
Operation test software: Delete from the parameter calculating charge.
Weak:
Operation test software: Delete weak bond from the parameter calculating.
Angles:
Operation test software: Remove the parameter from the calculation of bending and torsion angles.
Names:
Operation that allows the graphical display of the names associated with their atoms
Versors:
Operation that allows the graphical display of the unit vectors of the forces associated with their atoms.
orientations:
Operation that allows the graphical display of the orientations associated with their atoms.
Direction bond:
Operation that allows the graphical display of the bonds between their atoms, through the segments.
Torsional direction:
Operation that allows you to view the graphical the orientations torque between its atoms.
Link to run the simulation of proteins (Protein Folding) in a shortened version (JAVA)
Protein Dynamics Example
|


